")
CRISPR screening is supposed to accelerate discovery. But in too many labs, it does the opposite. All it takes is a batch of inconsistent viral titers, a poorly designed guide library, or one misstep in selection—and months of work disintegrate into unusable data.
And this isn’t rare. Many genome-scale CRISPR projects fail to yield actionable results. If you want to see what a validated CRISPR screen should actually look like, click here for a detailed walkthrough. Not because the science is flawed, but because small oversights cascade into systemic failures.
The good news? These failures are avoidable. In this article, we dissect the most common pitfalls in CRISPR screens, explain why traditional workflows often fall short, and explore how leading labs design smarter systems to produce reliable, reproducible data—fast.
Pitfall #1: Bad Guide Design Derails CRISPR Gene Editing
Every CRISPR gene editing screen starts with one essential component: a high-quality sgRNA library. But too often, teams use outdated or generic libraries that don’t match their experimental needs. These libraries may skip entire gene classes, lack isoform targeting, or rely on guide sequences with poor on-target efficiency.
The term “genome-wide” doesn’t guarantee comprehensive coverage. Without NGS-based validation, there’s no way to confirm guide distribution or sequence fidelity. Worse, if the library wasn’t designed for your cell type or condition, critical targets might be absent—or underrepresented.
Custom libraries sound like a fix, but designing them with outdated tools or skipping validation checks (like NGS readouts) just compounds the risk.
What smart labs do:
- Use libraries designed with up-to-date genome annotations.
- Validate guide coverage and diversity with NGS.
- Build redundancy into guide design (multiple sgRNAs per gene).
- Avoid “off-the-shelf” solutions unless thoroughly vetted.
Because if your guide library is flawed, everything downstream is already compromised.
Pitfall #2: Inconsistent Viral Delivery Wrecks Reproducibility
Even the best-designed library is useless if the sgRNAs don’t get into cells consistently. That’s why lentiviral delivery is a critical failure point in many screens.
Small fluctuations in viral titer or MOI (multiplicity of infection) can skew results. High MOI leads to multiple sgRNAs per cell, complicating analysis. Low MOI reduces representation, dropping statistical confidence. Worse, cell-type-specific responses to lentivirus are highly variable.
Poor viral purification can also introduce immune stress or toxicity, obscuring subtle phenotypes. If your delivery system introduces noise, the signal disappears.
What smart labs do:
- Use standardized, high-titer viral preps with batch QC.
- Validate MOI on each cell type before scaling.
- Prefer tool cell lines that express Cas9 consistently.
- Avoid “homebrew” virus whenever possible.
Consistency isn’t optional. In CRISPR gene editing, it’s the baseline for truth.
Pitfall #3: Skipping QC That Protects Data Integrity
Even with great libraries and delivery, screens can fail silently if quality checks are skipped. Assumptions like “our cells are fine” or “our virus is clean” have led countless screens into quiet disaster.
Without STR profiling, you might be using misidentified or contaminated cell lines. Without mycoplasma testing, invisible infections distort growth and response. Without checking guide representation pre- and post-transduction, your data could be biased beyond repair.
What smart labs do:
- Run STR profiling to confirm cell identity.
- Test all cell stocks for mycoplasma before experiments.
- Use viral titer quantification and sterility checks.
- Perform NGS before and after screening to confirm guide distribution.
This isn’t overkill. It’s insurance. In high-throughput experiments, the cost of missing a single check far outweighs the cost of doing it right.
Pitfall #4: No Functional Validation, No Real Insight
Just because an sgRNA shows enrichment or depletion doesn’t make it a true hit. Without downstream validation, your “discovery” could be a fluke.
Many screens stop at sequencing. But top labs know that functional follow-up is where hypotheses become findings. Did knocking out Gene X really drive the phenotype? Can reintroducing it reverse the effect? Is the pattern reproducible across guides and conditions?
What smart labs do:
- Follow up on hits with knockout/knock-in validation.
- Use orthogonal assays (e.g., protein expression, phenotypic rescue).
- Confirm reproducibility across sgRNAs and biological replicates.
Without this step, screens become noise machines. With it, they produce actionable insight.
Pitfall #5: Ignoring Data Analysis Best Practices
Even with perfect execution, weak data analysis can turn a strong experiment into a misleading conclusion. Without rigorous normalization, proper statistical controls, and batch effect correction, signal is easily lost or misinterpreted.
Common mistakes include:
- Misapplying models that ignore sgRNA variability
- Failing to account for sequencing depth or dropout events
- Relying solely on p-values without biological context
What smart labs do:
- Use CRISPR-specific bioinformatics tools (e.g., MAGeCK, PinAPL-Py)
- Cross-validate hits using multiple metrics and replicates
- Collaborate with informatics experts to avoid overfitting
Great CRISPR data isn’t just about what comes off the sequencer—it’s about what you do with it.
Smarter Systems: What Good CRISPR Workflows Actually Look Like
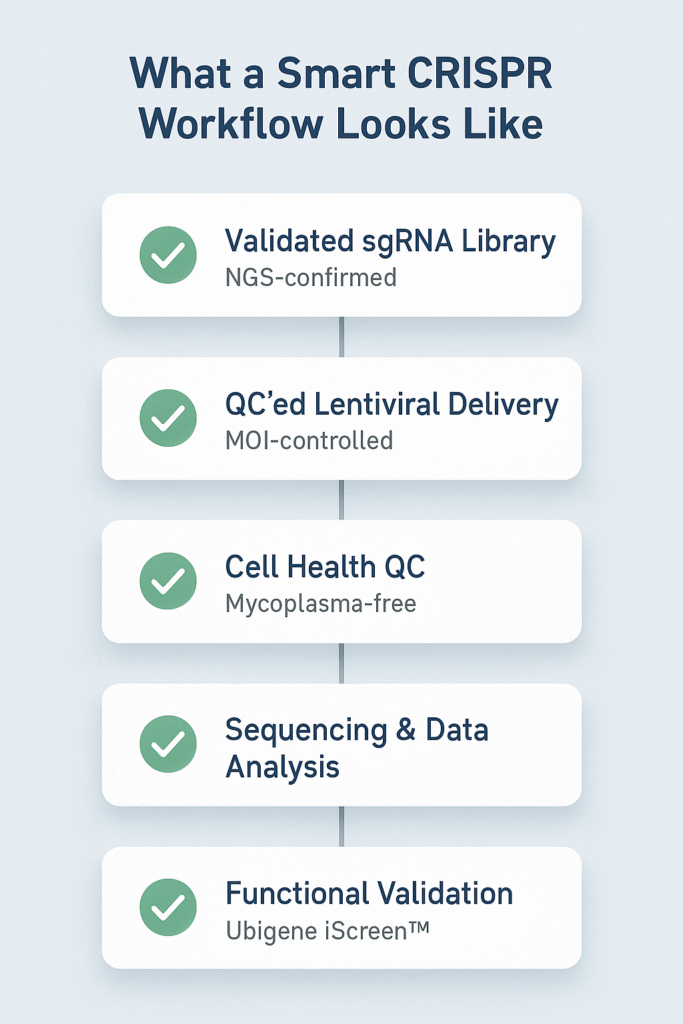
Avoiding these pitfalls isn’t just about doing more. It’s about doing it right—from day one. And increasingly, the best labs rely on integrated platforms that build reliability into the workflow itself.
Start with Validated Libraries:
- Full genome or pathway-targeted coverage.
- Optimized guides using current design tools.
- NGS-confirmed diversity and representation.
Standardize Delivery:
- High-titer, QC-certified lentivirus.
- Cell-line-specific MOI protocols.
- Validated Cas9-expressing tool lines where available.
QC at Every Stage:
- STR profiling for identity confirmation.
- Mycoplasma screening on all working stocks.
- Viral titer testing + contamination checks.
- Pre/post NGS validation of guide libraries.
Go Beyond Read Counts:
- Protein-level assays
- Phenotype rescue experiments
- Expert support for data interpretation
Platforms like Ubigene’s CRISPR-iScreen™ offer exactly this: validated guide design, standardized viral packaging, and integrated quality controls. It’s not about outsourcing—it’s about reducing the risk of failure.
Want a visual walkthrough of what a proper CRISPR screen looks like? This guide lays out the full workflow.
Conclusion: In CRISPR Screening, Failure Is Optional
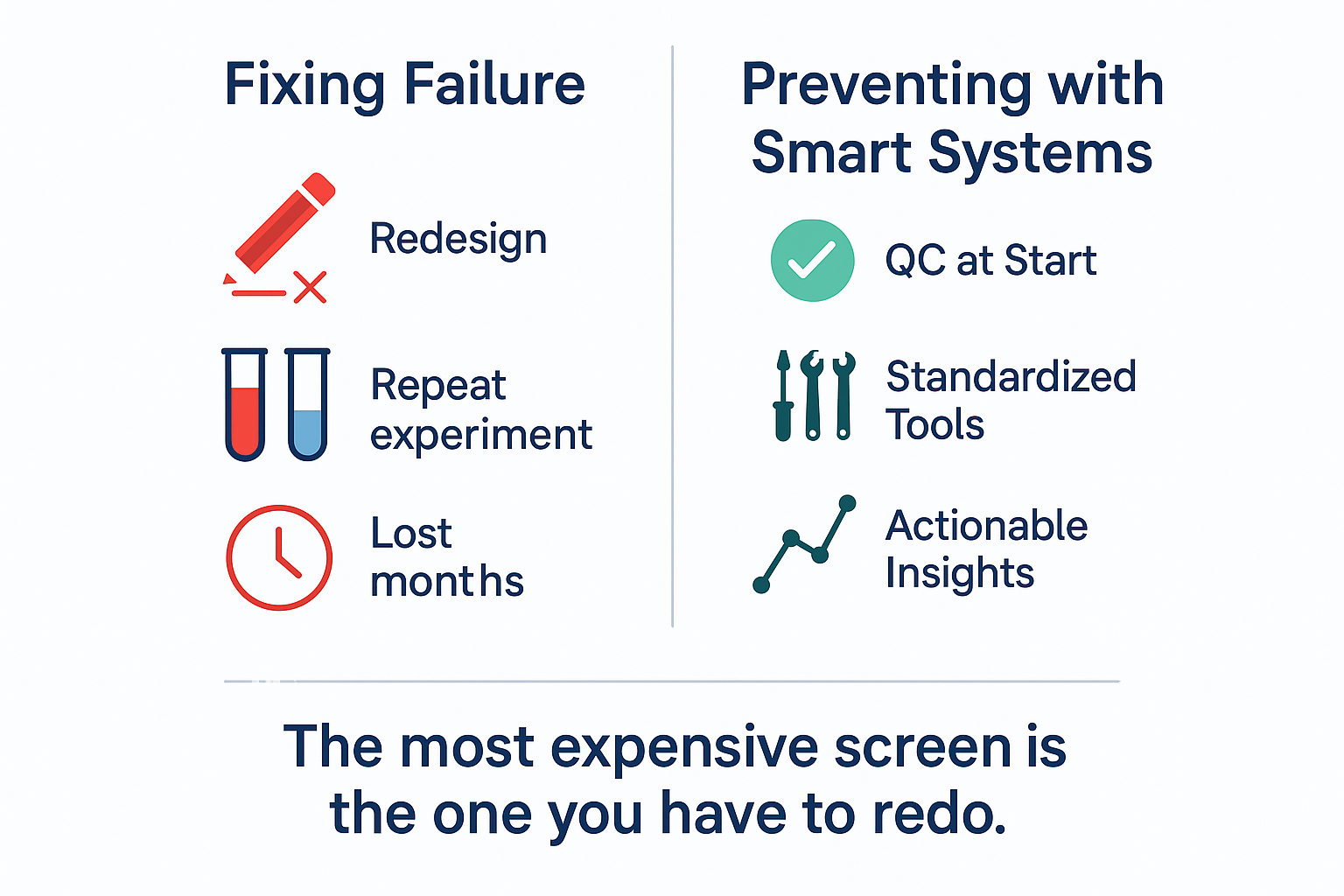
CRISPR technology can unlock powerful insights—but only when experiments work. The difference between a screen that reveals a new target and one that wastes six weeks often comes down to discipline, not discovery.
Don’t let subtle errors erase months of work. Build smart, systemized workflows that embed quality at every step. Whether you use an internal pipeline or partner with a platform like Ubigene, the message is the same:
You can’t afford a failed CRISPR screen. And the good news is—you don’t have to.